AT_genocon2021_b Practice 2
题目描述
[problemUrl]: https://atcoder.jp/contests/genocon2021/tasks/genocon2021_b
$s$ 和 $t$ 是仅由字母 A、C、G、T 组成的字符串。$s$ 和 $t$ 的第 $i$ 个字符分别记作 $s[i]$ 和 $t[i]$。现在可以在 $s$ 和 $t$ 的任意位置插入表示空白的字符 -,使得两者长度相同,并最大化被称为 Sum of pairs 的分数 $S_{sop}$。
$S_{sop} = \sum_{i=1}^{N} sim(s[i],\ t[i])$
其中,$N$ 是插入 - 后字符串的长度。$sim(x, y)$ 表示 $x$ 和 $y$ 的相似度,其输入输出如下表所示:
\[
\begin{array}{c|ccccc}
x \backslash y & A & C & G & T & - \\
\hline
A & 1 & -3 & -3 & -3 & -5 \\
C & -3 & 1 & -3 & -3 & -5 \\
G & -3 & -3 & 1 & -3 & -5 \\
T & -3 & -3 & -3 & 1 & -5 \\
- & -5 & -5 & -5 & -5 & -5 \\
\end{array}
\]
例如,若 $s = \text{CTCGGT}$,$t = \text{CATCC}$,在 $s$ 的第一个字符后插入一个 -,在 $t$ 的第五个字符后插入两个 -,则
$S_{sop} = sim(\text{C}, \text{C}) + sim(-, \text{A}) + sim(\text{T}, \text{T}) + sim(\text{C}, \text{C}) + sim(\text{G}, \text{C}) + sim(\text{G}, -) + sim(\text{T}, -) = 1 + (-5) + 1 + 1 + (-3) + (-5) + (-5) = -15$
此时 $S_{sop}$ 达到最大。将插入 - 后的 $s$ 和 $t$ 上下排列如下,可以看到上下同一位置尽量出现相同字符:
```
C-TCGGT
CATCC--
```
像这样,在字符串的适当位置插入空白 - 使两条字符串对齐,称为“配对比对”(pairwise alignment)。
请输出使 $S_{sop}$ 最大的两条输入字符串的配对比对。
输入格式
输入按以下格式从标准输入给出。
> $s$ $t$
输出格式
请输出使输入字符串的 $S_{sop}$ 最大的配对比对。按照示例,在 $s$ 和 $t$ 的适当位置插入 - 后,第一行输出 $s$,第二行输出 $t$。若存在多个使 $S_{sop}$ 最大的比对,任选其一输出即可。第一行和第二行的长度必须相同。并且,第一行去除所有 - 后应与 $s$ 完全一致,第二行去除所有 - 后应与 $t$ 完全一致。
说明/提示
### 限制
- $s$ 和 $t$ 均为仅由 A、C、G、T 组成的字符串。
- $10 \leq |s|, |t| \leq 400$($|x|$ 表示字符串 $x$ 的长度。)
### 问题背景
※ ***本项内容为出题背景,不影响解题,感兴趣者可阅读。***
配对比对在基因组序列分析的多个场景中都有应用。这里以个人基因组序列分析为例进行说明。
用于读取 DNA 基因组序列的装置称为测序仪(sequencer)。人类基因组全长高达 30 亿碱基,是非常长的序列,但测序仪无法一次性从头到尾读取整个序列。不同装置读取长度不同,短的可读取数百,长的可达数百万碱基的片段。因此,需要确定读取到的片段序列在基因组上的具体位置。为此,将读取到的片段与已知的人类标准基因组序列(称为参考基因组序列)进行比对,确定其位置。需要注意的是,读取到的片段序列与对应的参考基因组片段之间可能存在差异,原因有二:一是基因组序列因个体差异而略有不同,二是测序仪在读取时可能产生错误。这两种情况都会导致与参考基因组序列相比,出现如下差异:
替换(某个字符被另一个字符替换)
```
ATGC ← 参考基因组序列
ATCC ← 读取到的个人基因组序列
```
缺失(某个字符缺失)
```
ATGC ← 参考基因组序列
AT-C ← 读取到的个人基因组序列
```
插入(某个字符被插入)
```
ATC-C ← 参考基因组序列
ATCAC ← 读取到的个人基因组序列
```
为了将存在上述差异的序列的相似部分合理对齐,需要配对比对。需要注意的是,对于全长基因组和测序仪读取的片段等长度差异较大的序列进行比对时,需要采用多种技巧。此外,为获得良好的比对结果,相似度的设计也很重要。例如,在预计插入较少的情况下,应将 - 与其他字符的相似度设得更低。上述 $sim(x, y)$ 中,不同字符间(替换)的相似度相同,但也可以针对易发生替换的字符间设定更高的相似度等。
### 关于测序仪
如今能够轻松进行“个人基因组序列分析”,其实是最近才实现的。请看下图(引用自 NIH)。
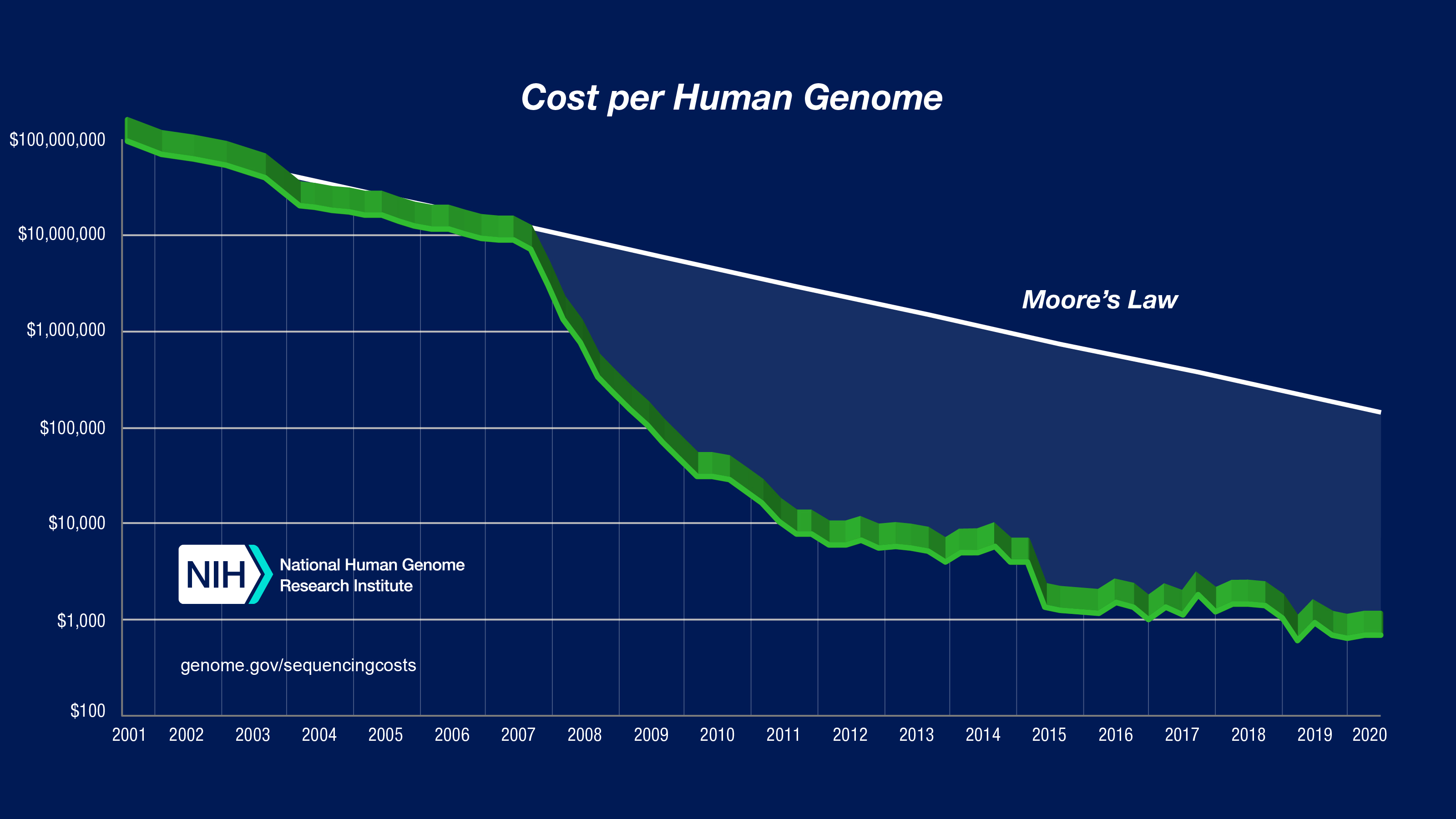
这是美国国立卫生研究院(National Institute of Health)估算的读取一人份人类基因组所需费用的变化图。纵轴为成本,横轴为年份。目前读取费用约为 10 万日元,而 20 年前竟需 100 亿日元。从图中可以看出,2000 年代后期成本急剧下降。究竟发生了什么?
实际上,正如前文所述,测序仪发生了技术革新,读取(测序)成本大幅下降。测序仪性能的提升极大推动了生命科学研究的发展,同时也极大提升了计算机科学在该领域的重要性。想想看,个人的“设计图”只需 10 万日元即可获得。全球研究机构产生了海量数据。每份设计图高达 30 亿碱基对,测序仪会以一定误差大量、零散地读取这些片段。没有优秀的算法,无法完成分析。
测序仪性能日新月异,相应地,解析其输出数据的算法也需不断进步。本题 D 所用数据即来自 [Oxford Nanopore Technologies](https://nanoporetech.com/) 的测序仪。该设备具有读取长度极长的优点,被广泛应用于各类分析场景。本次数据采用了最新的 Q20 套件,请大家关注。
由 ChatGPT 4.1 翻译